Introduction
Estrogens are perceived manly as ovarian sex hormones responsible for cellular proliferation and growth of tissues related to reproduction. In premenopausal women ovaries represent by far the most important source of circulating estrogens, although during pregnancy, placenta also secrets significant amounts of estrogens into the circulation (1). In addition to the effects on the female reproductive functions, estrogens also play a significant role in the regulation of skeletal homeostasis, lipid and carbohydrate metabolism, electrolyte balance, skin physiology, the cardiovascular system and the central nervous system (1,2). Due to this wide regulatory role, it is not surprising that estrogens also have an important function in male physiology and cannot be viewed solely as female sex hormones. Postmenopausal women, due to the decline in their ovary function, and men are largely dependent on local synthesis of estrogens in extragonadal target tissues. This local production of estrogens extends their signaling from endocrine to paracrine, autocrine and intracrine (3). The complexity is further increased by multiple mechanisms of estrogen signaling, namely the well established direct and indirect genomic signaling, the fast non-genomic actions of steroid hormones and ligand-independent signaling (4). Moreover, estrogen signaling is also tightly intertwined with epigenetic mechanisms which have been an important focus of research in recent years. Posttranslational histone modifications, microRNAs (miRNAs) and DNA methylation are important regulators of gene expression acting both upstream and downstream from estrogen receptors (ERs). Certain coregulators of estrogen signaling are able to induce posttranslational changes to histone proteins and specific epigenetic changes are necessary for initiation of transcription. Furthermore, different pathologies, aging and environmental factors can potentially influence estrogen signaling and regulation through induction of epigenetic changes (5,6). This review wishes to highlight additional aspects of estrogen signaling that exceed the classical concept of endocrine regulation and particularly point out the close relationship between estrogen signaling and epigenetic mechanisms.
Distribution of estrogen synthesis in the human organism
In addition to ovaries, several extragonadal tissues also produce estrogens. These include mesenchymal cells of the adipose tissue including that of the breast, osteoblasts and chondrocytes, aortic smooth muscle cells and vascular endothelium, as well as numerous parts of the brain (7). In contrast to the importance of ovaries in premenopausal women, in postmenopausal women and men extragonadal tissues represent both the predominant source of estrogen synthesis and the main site of estrogen action. Only estrogens that escape local metabolism can enter the circulation, suggesting that plasma levels of estrogens reflect rather than direct estrogen action in postmenopausal women and men (7). This was confirmed by several studies comparing tissue and plasma sex steroid concentrations. They showed that in postmenopausal women concentrations of estrogens in breast cancer and abdominal adipose tissue were several fold higher than in plasma (8-10). As a consequence, the levels of active steroid hormones in the circulation of postmenopausal women and men are probably of no significance in assessing their concentrations in individual extragonadal tissues (7). Sex- and menopause-based differences in estrogen synthesis are summarized in Table 1.
Table 1. Summary of sex- and menopause-based differences in estrogen synthesis (1,4,7,11,76).

In contrast to ovaries, estrogen production in extragonadal tissues is dependent on the availability of C19 steroid precursors, namely testosterone, androstenedione, dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS). In target tissues, testosterone can be converted to either 5α-dihydrotestosterone (DHT), the principal ligand for androgen receptors, or to 17β-estradiol, the most potent of the estrogens. Similarly, although requiring several enzymatic reactions, DHEAS, DHEA and androstenedione can also be converted to 17β-estradiol or DHT (Figure 1).
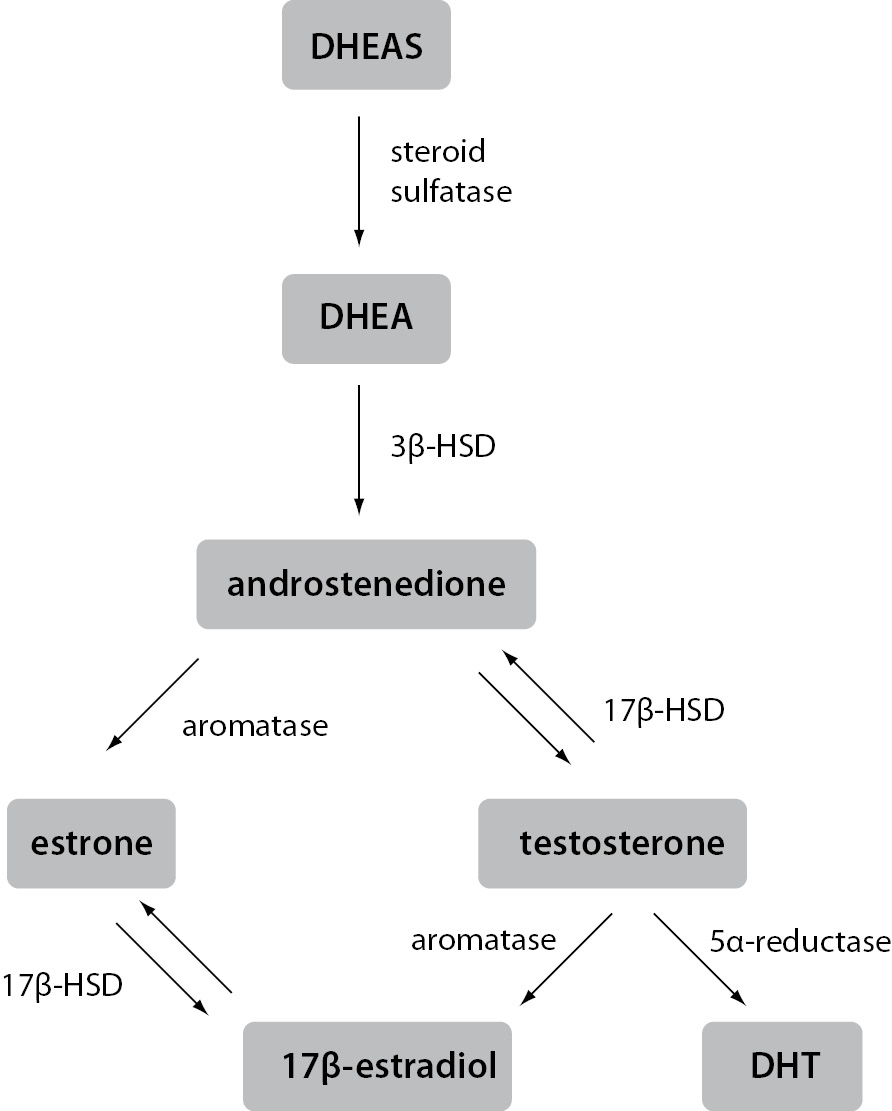
Figure 1. Schematic representation of estrogen synthesis in extragonadal tissues.
Estrogens and androgens are produced from C19 steroid precursors through several enzymatic conversions. Testosterone can be converted to the most active ligand on the androgen receptor, i.e. DHT, or the most active ligand on the estrogen receptor, i.e. 17β-estradiol, in a single reaction.
3β-HSD – 3β-hydroxysteroid dehydrogenase; 17β-HSD – 17β-hydroxysteroid dehydrogenas; DHEA – dehydroepiandrosterone; DHEAS – dehydroepiandrosterone sulfate; DHT – 5α-dihydrotestosterone
Distribution of the required enzymes varies in different extragonadal tissues, making the metabolism of DHEAS and DHEA tissue-specific. In plasma of postmenopausal women and men, DHEAS is by far the most abundant steroid precursor, followed by DHEA. Concentrations of both are several orders of magnitude greater than those of the active sex steroids, although their secretion and plasma concentrations decline significantly with advancing age. Interestingly, in postmenopausal women even the concentration of circulating testosterone exceeds that of 17β-estradiol by an order of magnitude. Approximately 25% of this testosterone is secreted directly from the ovaries, while the rest is formed in extragonadal tissues from steroid precursors. Androstenedione and DHEA are derived from both the adrenal cortex and the ovaries, whereas DHEAS is secreted entirely from the adrenals. The sheer amount of DHEAS and DHEA present in the circulation means they form a large reservoir of precursors available for the conversion to testosterone and estrogens in extragonadal tissues. Since testosterone can be converted in one reaction to either a ligand active at androgen receptor or a ligand active at estrogen receptors, it can be viewed as a circulating pro-hormone. Similar to the importance of estrogens in male physiology, high circulating concentrations of androgens indicate their significant role in women as well. In this light, it is impossible to separate estrogen signaling from androgen signaling or give them gender-specific roles. Therefore, it is always important to consider both regulatory pathways and their tight intertwinement in either women or men (3,7,11). This fact only further accentuates the central role of extragonadal production of steroid hormones, for a long time an overlooked aspect of estrogen signaling.
Structure and function of estrogen receptors
Estrogens exert their effects by binding to their designated receptors. In spite of the importance of estrogen signaling there are only three known receptors that mediate all of the estrogens´ effects. ERα was discovered first and is the most thoroughly investigated. Significantly homologues ERβ was identified almost four decades later and showed to have distinct, non-redundant roles (12). Another decade had to pass for the association of G protein-coupled estrogen receptor 1 (GPER1) with estrogen signaling. Characteristics of all three receptors are summarized in Table 2.
Table 2. Characteristics of estrogen receptors (2,12-15,17,18,77).
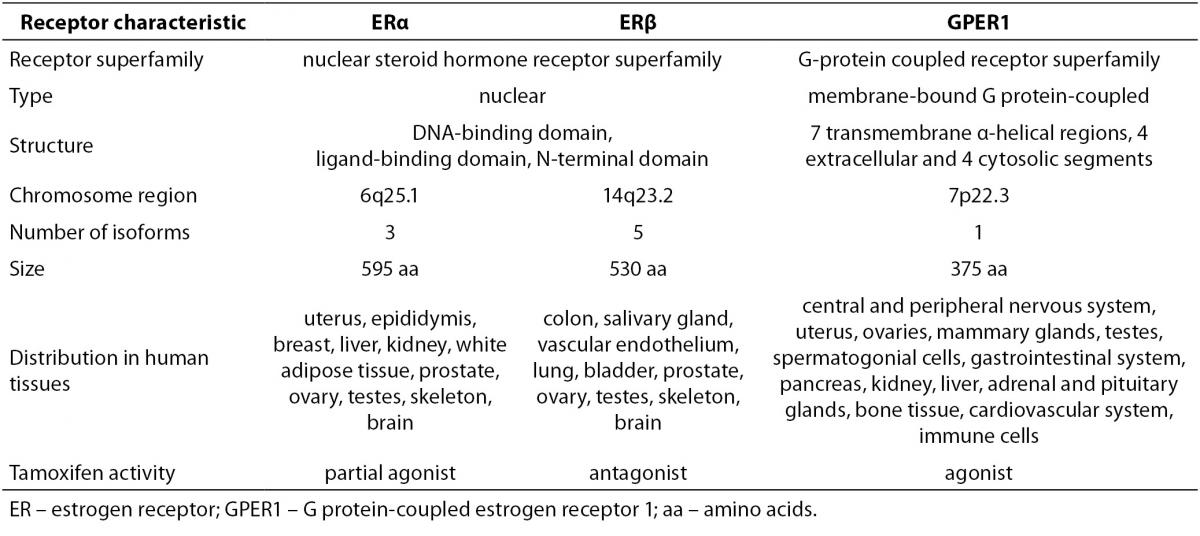
ERα has at least three and ERβ at least five different isoforms. Unlike the full length ERα, the two shorter ERα isoforms lack the N-terminal domain. Since they have the ability to heterodimerize with the full length ERα, they are able to repress ERα activity. The shortest ERα isoform termed ERα36, also acts as a membrane located ER able to interact with GPER1. The four shorter ERβ isoforms differ from the full length ERβ mostly in their ligand-binding domain, resulting in compromised ligand-binding ability. ERβ isoforms that are unable to bind ligands or coactivators and have no transcriptional activity dimerize preferentially with ERα, thereby silencing ERα signaling. This indicates that different ERα and ERβ isoforms can have a very diverse influence on estrogen signaling and target gene regulation (13,14). Furthermore, relative concentrations of ERα and ERβ in a given cell significantly influence its response to various ligands. Homodimers of ERα and ERβ were shown to regulate largely different sets of genes from that of ERα/β heterodimers. This is supported by the observation of completely distinct cellular responses by ER subtype-specific and non-specific agonists. It was also suggested that ERβ exerts an inhibitory effect on ERα-mediated signaling. Slightly different, in some instances even opposite, roles of ERα and ERβ are additionally underscored by differences in their expression in various tissues and organs (2).
GPER1, a plasma membrane receptor formerly known as an orphan G protein-coupled receptor 30 (GPR30), is genetically and structurally unrelated to ERα and ERβ. It is located on the cell surface and is responsible for rapid estrogen signaling, although certain ERα and ERβvariants have also been associated with the plasma membrane and the non-genomic signaling (13,14). GPER1 functions and is expressed independently from the two ERs. It has high affinity, limited capacity, displaceable, single binding sites for estrogens. In comparison to nuclear ERs, its binding affinity for 17β-estradiol is considerably lower and the rates of association and dissociation are very rapid and completed within a few minutes. It displays largely similar ligand binding specificities to those of the nuclear ERs with certain distinct differences. The specificity of GPER1 for estrogens is also underlined by the fact, that other steroid hormones possess very low binding affinities for this receptor (15-17).
Mechanisms of estrogen signaling
Estrogen dependent signaling can be roughly divided into genomic and non-genomic, based on the outcome of cellular events, e.g. modulation of gene expression or activation of signaling cascades, respectively. Furthermore, binding of the estrogen-ER complex to DNA can be either direct or indirect. Different mechanisms of estrogen signaling are represented in Figure 2.
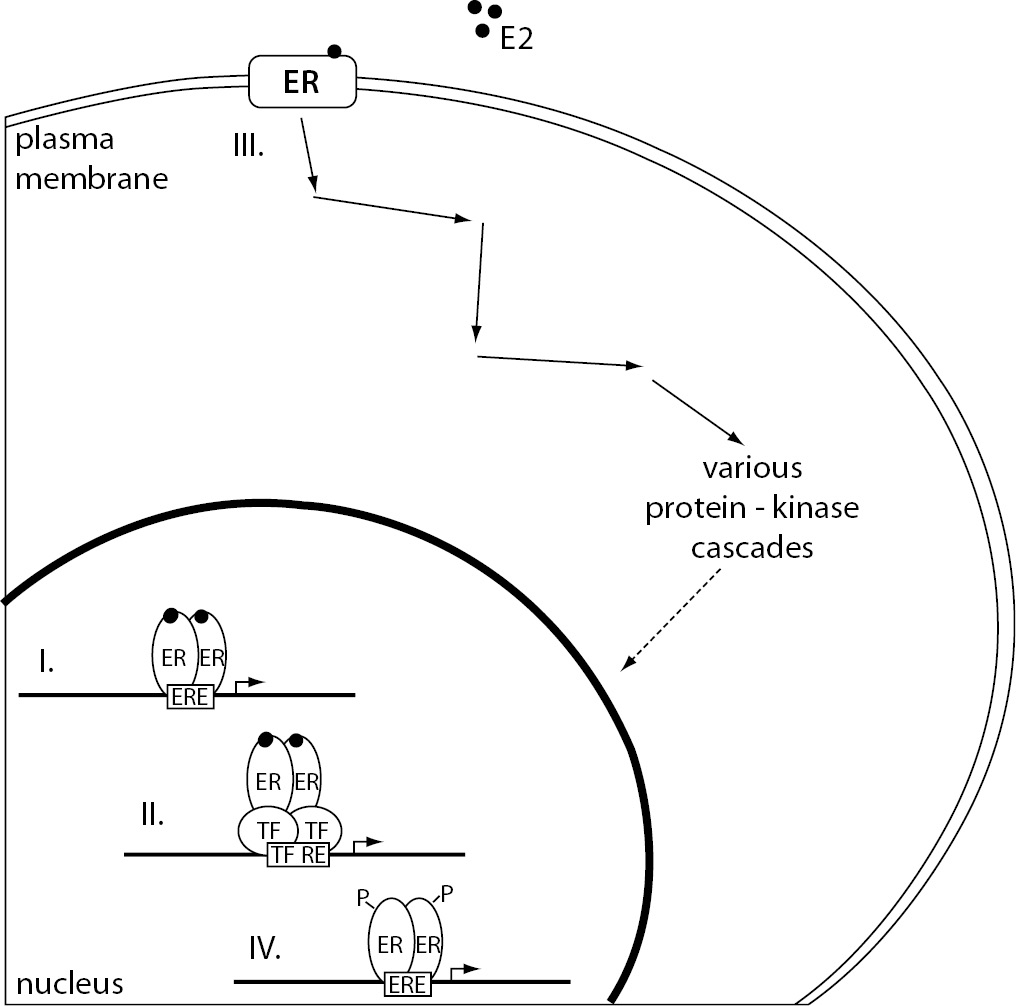
Figure 2. The representation of different mechanisms of estrogen signaling.
(I.) Direct genomic signaling pathway, considered the classical mechanism of estrogen signaling, promotes target gene expression by binding the E2-ER complex directly to the ERE. (II.) In the case of indirect genomic signaling pathway, E2-activated ERs bind DNA through protein-protein interactions with other classes of transcription factors at their respective response elements. (III.) Non-genomic signaling pathway starts with the binding of E2 to the ERs located at the plasma membrane resulting in the activation of various protein-kinase cascades. These can eventually lead to changes in gene expression due to phosphorylation of transcription factors. (IV). Ligand-independent signaling pathway causes ER activation and target gene transcription through phosphorylation of ERs or their associated coregulators.
E2 – 17β-estradiol; ER – estrogen receptor; ERE – estrogen response element; P – phosphate group; TF – transcription factor; TF RE – transcription factor response element.
Direct genomic signaling
Direct genomic signaling pathway is considered as the classical mechanism of estrogen signaling. The binding of 17β-estradiol to ERα or ERβin the cytoplasm of target cells causes conformational changes that enable receptor dimerization, translocation to the nucleus and binding to the estrogen response elements (EREs) located in or near the promoters of target genes. Binding of the ligand to the receptor also triggers recruitment of a variety of coregulators in a complex that alters chromatin structure and facilitates recruitment of the RNA polymerase II transcriptional machinery. In this way, estrogen-ER complex acts as a transcriptional activator promoting gene expression (18).
Indirect genomic signaling
17β-estradiol can also influence expression of genes that do not harbour EREs in their promoter regions. In fact, around one third of the estrogen responsive genes lack ERE-like sequences. In the case of ERE-independent genomic signaling ligand-activated ERs do not bind DNA directly, but rather through protein-protein interactions with other classes of transcription factors at their respective response elements. This mode of action enables activation or repression of target gene expression and significantly broadens estrogens´ regulatory influence. One of the best described examples includes interaction of estrogen-ER complex with FBJ murine osteosarcoma viral oncogene homolog (FOS) and jun proto-oncogene (JUN) proteins at the activator protein 1 (AP-1) binding sites in genes encoding ovalbumin, insulin-like growth factor 1 (IGF1), collagenase, cyclin D1 (CCND1) and choline acetyltransferase. The result depends on the ER subtype and type of the ligand. Other transcription factors that facilitate estrogen signaling also include Sp1 transcription factor, nuclear factor κB (NFκB), CCAAT/enhancer binding protein β (C/EBPβ), GATA binding protein 1 (GATA1) and signal transducer and activator of transcription 5 (STAT5) (19).
Non-genomic signaling
It has been known for a long time that certain estrogen-induced changes are simply too rapid to be associated with target gene transcription and subsequent protein synthesis. Non-genomic actions are common to steroid hormones and are usually associated with the activation of various protein-kinase cascades that can eventually lead to indirect changes in gene expression due to phosphorylation of transcription factors. Non-genomic estrogen signaling is most often associated with a subset of membrane bound ER, e.g. GPER1 and certain variants of ERα and ERβ. Binding of estrogens to ERs located at the cell surface can cause mobilization of intracellular calcium, stimulation of adenylate cyclase activity and cyclic adenosine monophosphate (cAMP) production, activation of the mitogen-activated protein kinase (MAPK) signaling pathway, activation of the phosphoinositol 3-kinase signaling pathway and activation of membrane tyrosine kinase receptors (14,19). GPER1 is specifically associated with the stimulation of adenylate cyclase and activation of EGFR (15). Molecular mechanisms underlying non-genomic estrogen signaling are without a doubt diverse and numerous and may depend on a number of conditions, like the availability of signal transduction molecules and downstream targets, suggesting a cell type-specific mechanism.
Ligand-independent signaling
In addition to genomic and non-genomic ligand-dependent estrogen signaling, ERs can also be activated in the absence of 17β-estradiol or another suitable ligand. Phosphorylation of the receptors on certain residues or their associated coregulators can cause ligand-independent ER activation. The two most often targeted amino acids are serine and tyrosine. Signaling pathways responsible for this modulation include regulators of general cellular phosphorylation state, such as protein kinase A (PKA) or protein kinase C (PKC), extracellular signals such as peptide growth factors, cytokines or neurotransmitters and cell cycle regulators. Peptide growth factors represent especially important group of estrogen-independent ER activators that include epidermal growth factor (EGF), insulin, IGF1 and transforming growth factor β (TGFβ). Principle intermediates between growth factors and ERs are the guanine nucleotide-binding protein p21ras and MAPK phosphorylation cascades, among others. Additional extracellular signals that modulate ER activity include heregulin, interleukin 2 and dopamine, as well as regulatory proteins cyclin A and D1 (12).
It has become clear that the classic genomic mechanism contributes only a small part to the complexity of estrogen signaling. It is expected that 17β-estradiol is able to regulate expression of the same target gene through multiple mechanisms, both genomic and non-genomic. In addition, the same promoter sequence can harbour both ERE as well as response elements associated with other transcription factors. The final gene response therefore depends on multiple factors including combination of transcription factors present on the gene promoter, expression levels and cellular localization of all three ERs, their numerous coregulators, and signaling components, as well as the nature of the stimuli. Since these variables can differ significantly among various cell types, it is possible that estrogens use distinct signaling pathways depending on the cellular context and in this way ensure very precise and cell-specific regulation of target gene expression.
Interactions of epigenetic mechanisms and estrogen signaling
Estrogen signaling controls several physiological processes by directly and indirectly regulating target gene transcription. Another group of important gene expression regulators receiving a lot of attention in recent years consists of epigenetic mechanisms. These include posttranslational histone modifications, miRNA and DNA methylation and are characterized by their ability to influence gene expression without altering the DNA sequence in a stable and potentially heritable manner.
Epigenetic mechanisms
Perhaps the most complex of the three, histone proteins can be subjected to various posttranslational modifications including acetylation, methylation, phosphorylation, ubiquitination, deimination and sumoylation. They are usually restricted to lysine and arginine residues but can also occur on serine and threonine. High levels of acetylation and trimethylated lysine (K) residue 4 of histone H3 (H3K4), H3K36 and H3K79 are associated with a more relaxed actively transcribed conformation of chromatin called euchromatin. Low levels of acetylation and high levels of H3K9, H3K27 and H4K20 methylation on the other hand result in a more condensed transcriptionally inactive heterochromatin. Most posttranslational histone modifications are dynamic and are regulated by families of enzymes that either bind or remove specific functional groups. For example, histone acetyltransferases (HATs) add, while histone deacetylases (HDACs) remove acetylation marks. Histone methyltransferases add methyl groups to arginine and lysine residues, while deiminases and lysine demethylases reverse these changes. There is also a similar relationship between histone kinases and phosphatases (20,21).
miRNAs perform their regulatory role in a very different way. They are small, approximately 21 nucleotide-long, non-coding RNAs usually associated with target gene repression. They facilitate binding of the RNA-induced silencing complex (RISC) to the target mRNA, usually within its 3´ UTR region. When miRNA sequence completely matches that of the target, mRNA is degraded. More often, miRNAs bind imperfectly to their targets, resulting in translational inhibition without mRNA cleavage. This can result in decreased target protein levels without any changes in the mRNA concentration. Individual miRNAs generally regulate multiple target genes at modest levels and many different miRNAs can target the same transcript (22).
Similar to miRNA-mediated posttranscriptional regulation, DNA methylation is mainly associated with gene repression. It is a reversible covalent binding of a single methyl group to the 5´ carbon of a cytosine residue located next to a guanosine. These dinucleotides commonly annotated as CpGs, occur with higher than expected frequency in regions referred to as CpG islands. These are unevenly distributed throughout the genome, located in promoter regions of genes, within genes and in intergenic locations. DNA methylation patterns are established by three DNA methyltransferases (DNMTs), namely DNMT1, DNMT3A and DNMT3B, and can be preserved during DNA replication and mitosis. This enables the inheritance of DNA methylation-mediated repression of target gene expression. The mechanisms of DNA demethylation which can be either passive or active are not yet fully elucidated (20).
Coregulators of estrogen signaling
As already mentioned, recruitment of a variety of coregulators in large multifunctional protein complexes is intrinsic to estrogen signaling. Most of these coregulators interact with many members of the nuclear receptor superfamily and are involved in chromatin remodeling, histone modifications, transcription initiation and elongation, splicing and proteasomal degradation. In addition to gene activation through coactivator recruitment, estrogen signaling is also associated with repression of transcription assisted by corepressors (12,13). Coactivators associated with estrogen-mediated transcription include v-src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC1), nuclear receptor coactivator 2 (NCOA2), NCOA3, proline, glutamate and leucine rich protein 1 (PELP1), CREB binding protein (CBP), E1A binding protein p300 (p300), K(lysine) acetyltransferase 2B (PCAF/KAT2B), coactivator-associated arginine methyltransferase 1 (CARM1) and protein arginine methyltransferase 1 (PRMT1) while estrogen-induced repression recruits corepressors nuclear receptor corepressor 1 (NCOR1), NCOR2 and metastasis associated 1 (MTA1) among others (23). Even though coregulators are involved in a multitude of transcriptional steps we would like to point out their role as intermediates between transcriptional coregulation and epigenetic changes. A number of coregulators possess chromatin-modifying activities such as acetyltransferase and methyltransferase activity. In fact, histone acetylation is a necessary early step in transcription initiation marking active chromatin sites. Acetylation, as well as certain methylation marks attract additional transcriptional coactivators which in turn recruit general transcriptional machinery, including RNA polymerase and its accessory factors. The most commonly utilized HATs include p300, CBP, PCAF, KAT2A and KAT5. Conversely, removal of these acetylation marks from histones by corepressor complexes is associated with target gene silencing. Common HDACs include sirtuin 1 (SIRT1) and HDAC1-4 (24). Clearly chromatin modifications are integral to estrogen-mediated transcriptional regulation and gene expression in general.
Epigenetic regulation of estrogen signaling
Another contact point between estrogen signaling and epigenetic mechanisms is the epigenetic control of ER gene expression. Through the regulation of ER levels the epigenetic control extends to all ER target genes and estrogen-regulated processes. Several studies have shown that histone-modifying enzymes like, e.g. histone deacetylases HDAC1 and HDAC3 and histone methyltransferase suppressor of variegation 3-9 homolog 1 (Drosophila) (SUV39H1), are at least in part responsible for the reduction of ERα expression in oncological settings, specifically in breast cancer (25-29). Conversely, histone deacetylase SIRT1 activity is necessary for ERα expression in breast cancer. However, it was shown that SIRT1 is not directly associated with the ERα promoter and does not participate in histone modifications but is rather involved in the formation of basal transcription factor complexes at the ERα promoter. In addition, ERα and SIRT1 proteins directly interact to form transcriptional activator or repressor complexes depending on the targeted gene. This indicates that SIRT1 has other regulatory roles in addition to histone deacetylation (29,30). The balance between HATs and HDACs present at ERα gene promoter was also pointed out as an important determinant of gene expression and repression. Furthermore, it was suggested that histone-modifying enzymes establish a more transient gene repression followed by the recruitment of DNMTs to the same regulatory complexes in order to methylate DNA for long-term gene silencing (26,31). Despite the lack of data on direct associations between different histone modifications and ERβgene expression, several studies reported re-expression of ERβ after treatment with trichostatin A (TSA), an HDAC inhibitor, in breast, ovarian and prostate cancer cell lines. This effect was further increased when a DNMT inhibitor, 5-aza-2’-deoxycytidine (5-AZA), was added to the TSA treatment (32-35). This supports the observed complementary roles of histone deacetylation and DNA methylation in repressing gene expression. An increase in ERβ expression was also observed in a non-oncological setting after exposure to sodium butyrate, another HDAC inhibitor (36).
Similarly, miRNAs were also identified as important regulators of ER gene expression. ERα was shown to be directly targeted by numerous miRNAs, including miR-18a, miR-22, and miR-206 (for a more comprehensive list see Table 3) (37-45). There is markedly less information available about miRNAs targeting ERβ and GPER1. So far only miR-92 was identified as a miRNA that directly targets ERβ, while no such miRNAs are known for GPER1 (22, 46).
Table 3. Experimentally validated miRNAs that directly regulate ER gene expression.

DNA methylation is probably the most thoroughly analyzed epigenetic mechanism responsible for ER silencing in numerous malignancies. As already indicated, expression of several ERα and ERβ isoforms was shown to be inversely correlated with the methylation of CpGs at their respective promoters. This association was observed repeatedly in multiple cell lines, primary cell cultures and tissue samples of hematopoietic neoplasms, endometrial, prostate, breast, and ovarian cancers, among others (47-53).
Even though the described studies focused mainly on epigenetic changes that resulted in ER silencing in numerous malignancies, they prove beyond a reasonable doubt that epigenetic mechanisms play an important role in the regulation of ER expression and consequently in the regulation of estrogen signaling.
Estrogen-mediated control of epigenetic mechanisms
The classic view of estrogen receptors as transcription factors suggests that the roles of estrogen signaling and epigenetic regulation can also be reversed, meaning that estrogen signaling is also able to regulate expression of certain chromatin-modifying enzymes and miRNAs (54).
An in vivo study showed a stimulatory effect of estrogen replacement on SIRT1 protein levels in bone marrow of estrogen-deficient mice (55). Activation of SIRT1 transcription by 17β-estradiol through ERα was also shown in breast cancer cell lines (29). Similarly, HDAC6 expression is also positively regulated by estrogen signaling (56). Furthermore, a transcriptome profiling study in primary osteoblasts identified lysine (K)-specific methyltransferase 2D (MLL2) as an estrogen-responsive gene (57). These examples clearly show that estrogen signaling plays an important role in the regulation of certain histone-modifying enzymes.
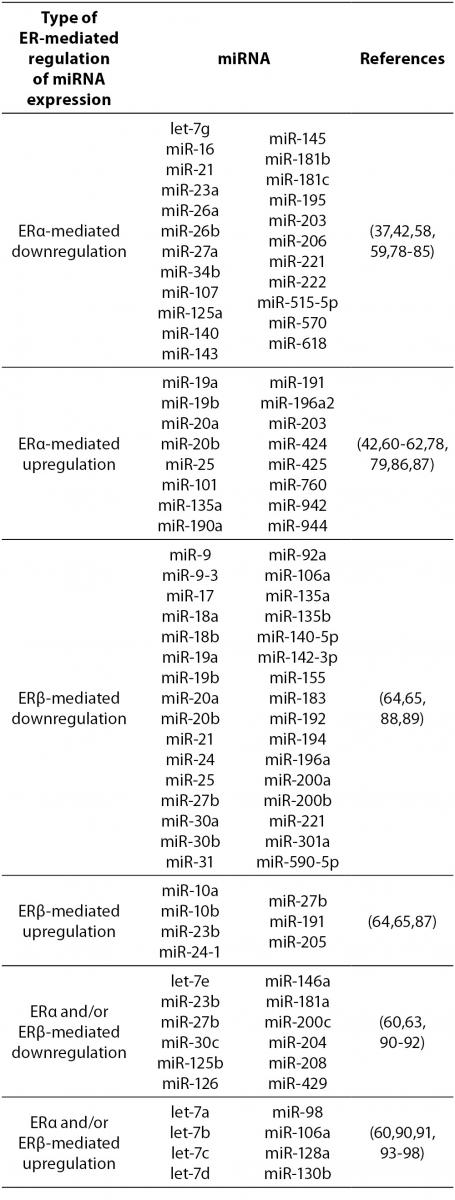
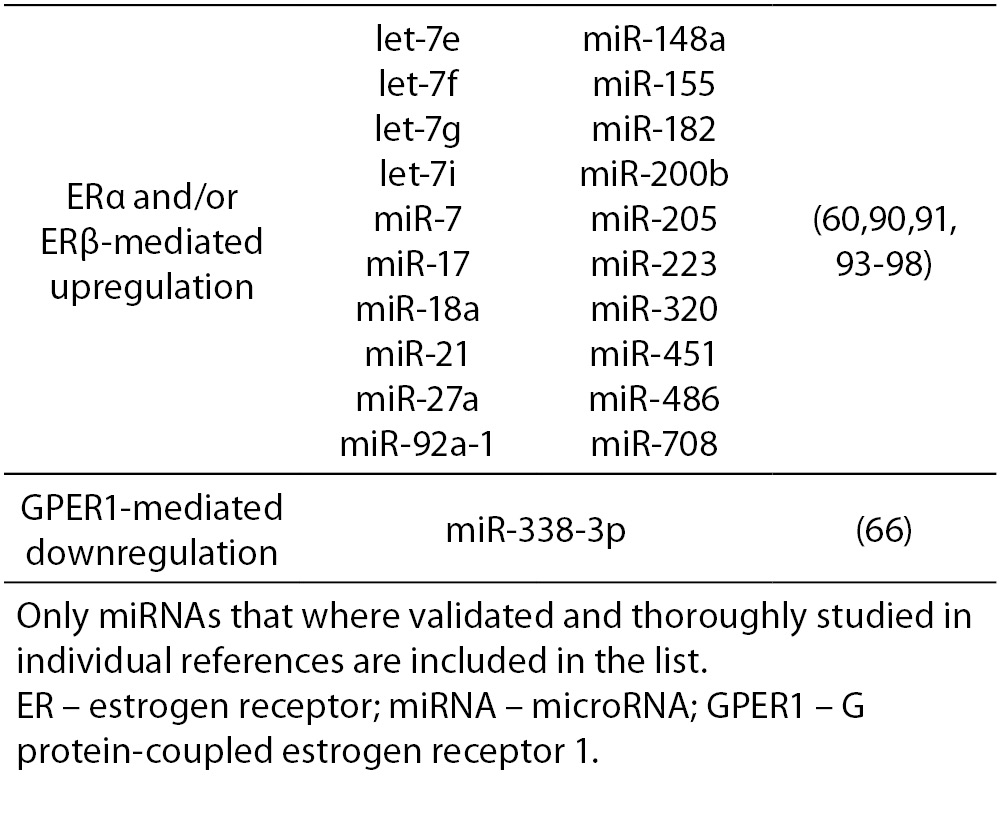
17β-estradiol also significantly upregulates and downregulates expression of numerous miRNAs. All three ERs were shown to directly and indirectly influence transcription of miRNAs in tumorous and normal cells. ERα-mediated estrogen signaling inhibits transcription of miR-21, miR-26a, miR-140, miR-181b and miR-206, while it stimulates expression of miR-190a, miR-191, miR-203 and miR-425 among others (37,58-63). ERβ is on the other hand associated with downregulation of miR-17, miR-30a, miR-200a and miR-200b, and upregulation of miR-23b, miR-24-1 and miR-27b among others (64, 65). Additionally, binding of 17β-estradiol specifically to GPER1 was shown to repress the expression of miR-338-3p (66). A more comprehensive list of ER-regulated miRNAs is presented in Table 4.
Table 4. ER-mediated regulation of miRNA expression.
As mentioned earlier, DNMTs are responsible for maintenance and de novo methylation of DNA. Estrogen signaling was clearly shown to influence the expression of DNMTs, although the results are quite diverse. DNMT1 was shown to be either downregulated or unaffected by 17β-estradiol, DNMT3A was downregulated, upregulated or unaffected in the same conditions, while DNMT3B could be downregulated or upregulated. There are many potential explanations for this discordance, the most obvious being differences between various malignant and non-malignant cell lines used by researchers, differences in their ER levels and differences in the signaling coregulators present (14,67-72). In line with this, studies observed that estrogen signaling either increased DNA methylation and subsequently epigenetically silenced target genes or predominantly demethylated DNA resulting in epigenetic upregulation of downstream targets (31,67,68,73). Furthermore, developmental exposure to relevant doses of 17β-estradiol or xenoestrogens induced hypomethylation of certain and hypermethylation of other genes, increasing the susceptibility to certain types of adult-onset cancer (72,74,75).
Estrogen signaling end epigenetic regulation are two ubiquitous regulatory mechanisms essential for maintaining cell and tissue homeostasis. Due to their importance it is not surprising that their functions tightly intertwine. In addition, their complementary roles increase the level of cell-specificity and fine-tuning of transcriptional regulation.
Conclusions
In the last two decades, we saw a dramatic increase in our understanding of estrogen signaling. Extragonadal synthesis of estrogens and their tight connection with androgens have extended the mechanism of estrogen action from endocrine to paracrine, autocrine and even intracrine signaling. Estrogens and androgens have lost their gender-limited roles importantly contributing to the physiology of several tissues and organ systems. Furthermore, at least three different estrogen receptors, namely ERα, ERβ and GPER1, with distinct, non-redundant and in certain cases opposite roles are now known. Indirect genomic signaling through the interactions with other transcription factors at their DNA response elements and the fast non-genomic signaling mediated by membrane-bound ERs additionally increase the complexity of estrogen signaling. Importantly, estrogen signaling is also tightly connected to epigenetic mechanisms both as the regulator of gene expression of certain chromatin-modifying enzymes and miRNAs, as well as their target through the epigenetic regulation of ER gene expression levels. Moreover, certain epigenetic changes are intrinsic to estrogen-mediated transcriptional regulation and are executed at least in part by coregulators of estrogen signaling. Epigenetic mechanisms thus augment not only the intricacy but also the specificity and fine-tuning of estrogen-mediated transcriptional control. Better comprehension of the complexity of estrogen regulation has deepened our understanding of the effects of the existing estrogen receptor modulators as well as opened opportunities for the discovery of new tissue- and perhaps even cell-specific compounds.
Acknowledgments
This work was supported by the Slovenian Research Agency (grants ARMR19, P3-0298, J3-2330 and J3-5511).