Introduction
UDP-glucuronosyltransferase 1A1 (UGT1A1) is the only physiologically relevant enzyme for bilirubin glucuronidation in humans (1). Sequence variations in the UGT1A1 encoding gene (UGT1A1) that decrease the enzyme activity are responsible for inherited unconjugated hyperbilirubinemias ranging from Gilbert's syndrome (GS) (30% activity) to Crigler-Najjar syndrome type II (10% activity) and Crigler-Najjar syndrome type I (0% activity) (2,3). While both Crigler-Najjar syndromes are very rare, GS is quite common (4). On the basis of serum bilirubin concentration, 3-10% of the general population are estimated to have GS. The condition is characterised by intermittent mild unconjugated hyperbilirubinemia in the absence of hemolysis or evidence of liver disease (2,5). The causative sequence variation in Caucasians is almost exclusively a (TA) dinucleotide insertion in the TATA-box of the UGT1A1 promoter. Affected individuals are homozygous for the variant promoter and have 7 TA repeats instead of 6 TA repeats (6). The frequency of (TA)7/7 genotype in Caucasians is 11-16% (4). Promoters with 5 and 8 TA repeats have also been identified; first in individuals of African descent and subsequently in Caucasians, where they are extremely rare (7-11). An increase in promoter length results in a decrease in enzyme transcription and therefore decreased conjugation of bilirubin (7,8,12). Although GS is quite benign from a hepatological point of view, screening for this syndrome is important clinically in the differential diagnosis of jaundice and because these subjects may be more susceptible to the adverse effects of several drugs that are metabolised by UGT1A1, such as irinotecan (9,31). In addition, the UGT1A1(TA)n polymorphism has been associated with neonatal hyperbilirubinemia, hyperbilirubinemia and an increased likelihood of gallstone formation in several inherited haematological disorders and in cystic fibrosis (4,10).
Several methods for genotyping the UGT1A1(TA)n polymorphism based on differentiation according to the length, conformation or melting behaviour of the fragments have been reported. They differ with respect to throughput, cost, specificity and sensitivity, as well as the manual work required, time, special instruments and other characteristics. Besides sequencing with fluorescence-based dideoxy chain terminator method, different fragment analyses, the Invader® assay, real-time fluorescent PCR using hydrolysis or hybridization probes, pyrosequencing, single strand conformation polymorphism (SSCP), high resolution melting and denaturing high performance liquid cromatography (DHPLC) analyses have been described (8-10,14-19). DHPLC is commonly used to identify single nucleotide substitutions as well as small insertions and deletions for mutation detection and genotyping under partially denaturing conditions (20). To much lesser extent, the DHPLC sizing method can also be applied for genotyping (21).
The two published DHPLC assays enable separation of the three common genotypes (TA(6/6), TA(6/7) and TA(7/7)) using either partially denaturing or fully denaturing conditions. However, no protocols for simultaneous genotyping of common and rare genotypes of the UGT1A1(TA)n polymorphism using DHPLC method have been described so far.
In this study we present a new sizing DHPLC assay for simultaneous genotyping of common and rare UGT1A1(TA)n variants and compare it with the previously described SSCP assay.
Materials and methods
Materials
Fifty-seven DNA samples, which have been previously genotyped by SSCP assay were included in the study. Genomic DNA was isolated from peripheral blood leukocytes using FlexiGene DNA kit (Qiagen, Hilden, Germany). The study was conducted according to the Declaration of Helsinki and approved by the Ethical Committee of the Republic of Slovenia, No. 33/05/04. Subjects’ written informed consents were obtained before entering the study.
Methods
Genotyping of UGT1A1(TA)n promoter polymorphism by SSCP assay
The number of TA repeats was determined by polymerase chain reaction (PCR) followed by single-strand conformation polymorphism analysis (SSCP) as previously described (10) Briefly, UGT1A1 promoter fragments 199-205 bp in length, depending on the number of TA repeats, were amplified by PCR with primers F: 5'-TGAAATTCCAGCCAGTTCAA-3' and R: 5'-AGAGGTTCGCCCTCTCCTAC-3'. SSCP analysis was run on 8% (37:1) polyacrylamide gels in a Protean II electrophoresis unit (Bio-Rad Laboratories Inc., Hercules, CA) in 0.5X TBE buffer (50 mM Tris-borate, pH 8.3 and 0.5 mM EDTA) at a constant power of 20 watts at 9 °C for 3.5 h. After electrophoresis SSCP patterns were visualized by silver staining. The genotype of PCR products corresponding to each SSCP pattern were determined by sequencing, performed by MWG Biotech AG (Ebersberg, Germany) with the Value Read Service, applying the fluorescence-based dideoxy chain terminator method.
Genotypes were assigned as follows:
- (TA)5/6 (heterozygote with 5 TA and 6 TA repeats);
- (TA)5/7 (heterozygote with 5 TA and 7 TA repeats);
- (TA)6/6 (homozygote with 6 TA repeats);
- (TA)6/7 (heterozygote with 6 TA and 7 TA repeats);
- (TA)7/7 (homozygote with 7 TA repeats);
- (TA)6/8 (heterozygote with 6 TA and 8 TA repeats); and
- (TA)7/8 heterozygote with 7 TA and 8 TA repeats).
Genotyping of UGT1A1(TA)n promoter polymorphism by DHPLC assay
For DHPLC analysis a new pair of primers was designed for amplification of UGT1A1 promoter fragments 71-77 bp in length, depending on the number of TA repeats. The sequences of primers were as F: 5‘-CACAGTCAAACATTAACTTGGTG-3‘, R:
5‘-GTTCGCCCTCTCCTACTT-3‘. Oligonucleotide primers were designed based on the sequence of the UGT1A1 gene available in Genebank (accession no. AF352795.1) using mutationdiscovery.com. The PCR reaction mixture (25 μL) contained genomic DNA (100 ng), 1X PCR buffer, 0.2 mM each of the four deoxyribonucleotides, 2.0 mM MgCl2, 0.25 mM each of the two oligonucleotide primers and 0.5 units of AmpliTaq GoldTM polymerase (Applied Biosystems, Foster City, CA). Cycling conditions consisted of an initial 10 min at 95 °C, followed by 38 cycles of 30 s at 95 °C, 30 s at 57 °C and 20 s at 72 °C, and a final 7 min at 72 °C. Aliquots of PCR products were electrophoresed on 2% agarose gel to check their quality and quantity.
5‘-GTTCGCCCTCTCCTACTT-3‘. Oligonucleotide primers were designed based on the sequence of the UGT1A1 gene available in Genebank (accession no. AF352795.1) using mutationdiscovery.com. The PCR reaction mixture (25 μL) contained genomic DNA (100 ng), 1X PCR buffer, 0.2 mM each of the four deoxyribonucleotides, 2.0 mM MgCl2, 0.25 mM each of the two oligonucleotide primers and 0.5 units of AmpliTaq GoldTM polymerase (Applied Biosystems, Foster City, CA). Cycling conditions consisted of an initial 10 min at 95 °C, followed by 38 cycles of 30 s at 95 °C, 30 s at 57 °C and 20 s at 72 °C, and a final 7 min at 72 °C. Aliquots of PCR products were electrophoresed on 2% agarose gel to check their quality and quantity.
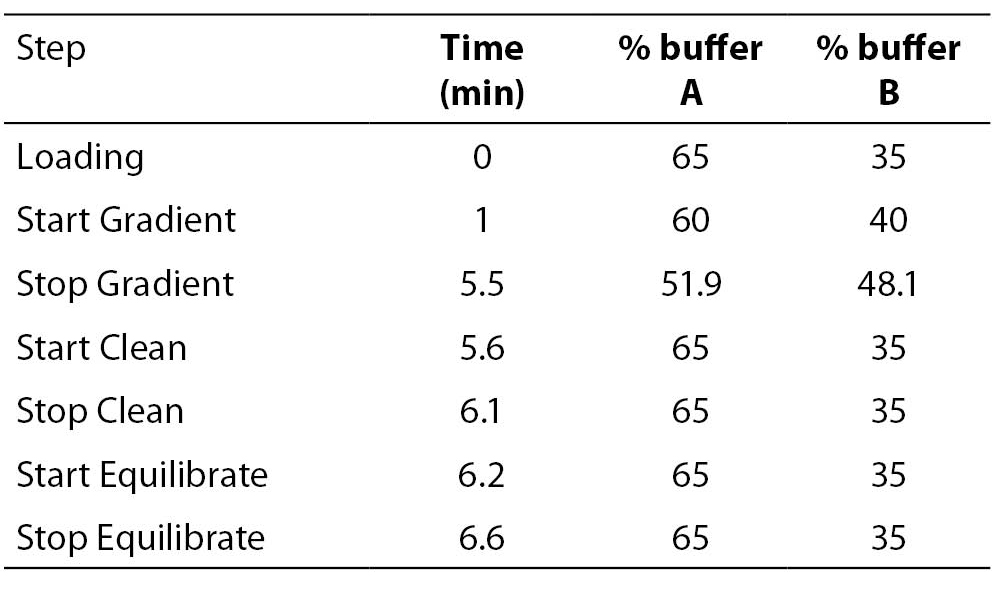
8 μl of the PCR products were further injected onto a preheated reverse-phase column (DNASep Column, Transgenomic Ltd, Omaha, USA) of a WAVE MD DHPLC system (Transgenomic Ltd, Omaha, USA) equilibrated by an ion pairing agent TEAA 0.1 M (Triethylammonium acetate). UGT1A1 promoter (TA)n repeat polymorphism was determined using a sizing mode by a linear acetonitrile gradient, achieved by mixing a buffer A (TEAA 0.1 M) with a buffer B (TEAA 0.1 M and acetonitrile 25%) with 1.8% per minute gradient increase from the start gradient of 40% up to stop gradient of 48.1% of the buffer B at constant temperature 50 °C and constant flow rate of 0.9 mL/min. The eluted DNA was detected at 260 nm. Genotypes of 57 individual PCRs were determined using the sizing calling routine (Transgenomic Navigator Software 1.6.2.). Details of the DHPLC protocol are listed in Table 1.
Table 1. DHPLC protocol using sizing mode
Results
Genotype distribution, sample errors and time of analysis using DHPLC and SSCP assays
Fifty-seven samples were genotyped for the UGT1A1(TA)n polymorphism twice, using DHPLC and SSCP assays. The genotype distribution in our selected study group is presented in Table 2.
Table 2. Distribution of UGT1A1(TA)n genotypes determined by SSCP and DHPLC assays
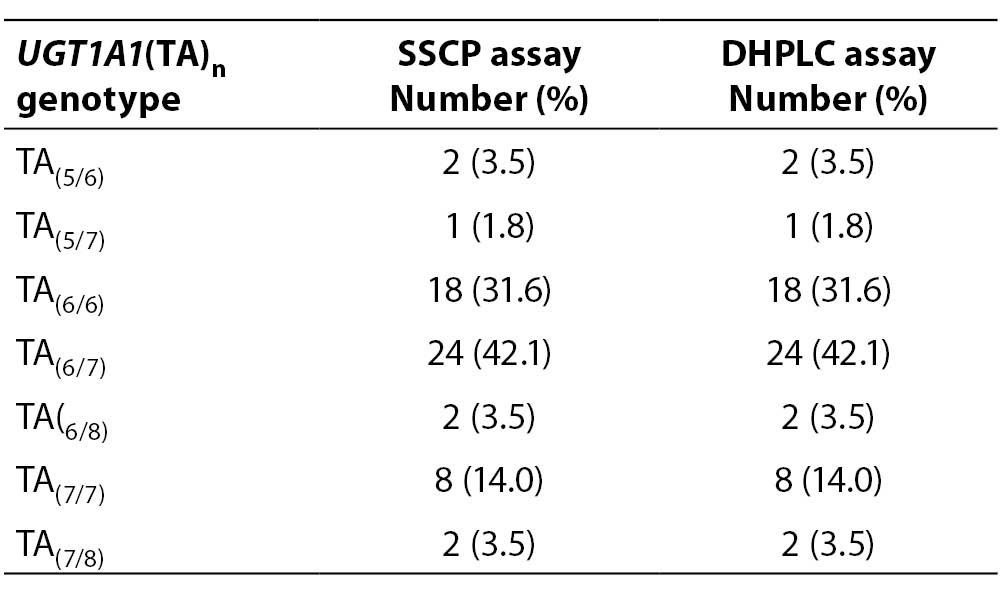
Our study group was absent of (TA)8/8 and (TA)5/5 homozygotes and (TA)5/8 heterozygotes. Most common genotypes were (TA)6/7 and (TA)6/6. When comparing the genotype results for each sample obtained by SSCP and DHPLC, no sample genotype error was detected (0%).
To achieve efficient discrimination between peaks of each UGT1A1(TA)n genotype we decreased the length of PCR amplicon (from 199-205 bp for SSCP to 71-77 bp for DHPLC analysis) and gradient increase of buffer B in DHPLC column was set up to only 1.8% per minute. The gradient increase finished in 48.1% of buffer B with the aim to shorten the retention time to only 6.6 minutes. The 96-well plate with 57 samples was analyzed within cca 6 h and 30 min using DHPLC, whereas the duration of analysis of the same number of samples was cca 2 working-days using SSCP assay.
Allelic discrimination of the common UGT1A1(TA)n polymorphism genotypes
The sample with (TA)6/7 genotype was used as a quality control for determining the common genotypes of UGT1A1 promoter polymorphism such as (TA)7/7, (TA)6/6 and (TA)6/7. Well-resolved separation of the chromatogram peaks between all three genotypes was observed. Allelic size discrimination of UGT1A1 promoter (TA)6/6, (TA)6/7 and (TA)7/7 repeat genotypes using DHPLC assay is presented in Figure 1. Samples with 6 TA and 7 TA repeat alleles eluted from the column by showing the peaks at a retention time ranges of 4.244-4.267 min and 4.408-4.428 min, respectively. The early eluting (TA)6/6 and later eluting (TA)7/7 genotype peaks showed 2-times higher peaks compared to peaks of heterozygotes ((TA)6/7) with additional small peak eluted earlier. The peaks of (TA)6/7 heterozygotes were double having the same retention times for 6 and 7 TA repeat alleles as of the homozygotes (TA)6/6 and (TA)7/7. The time shifting was very slight or absent at the same column pressure and buffer conditions.
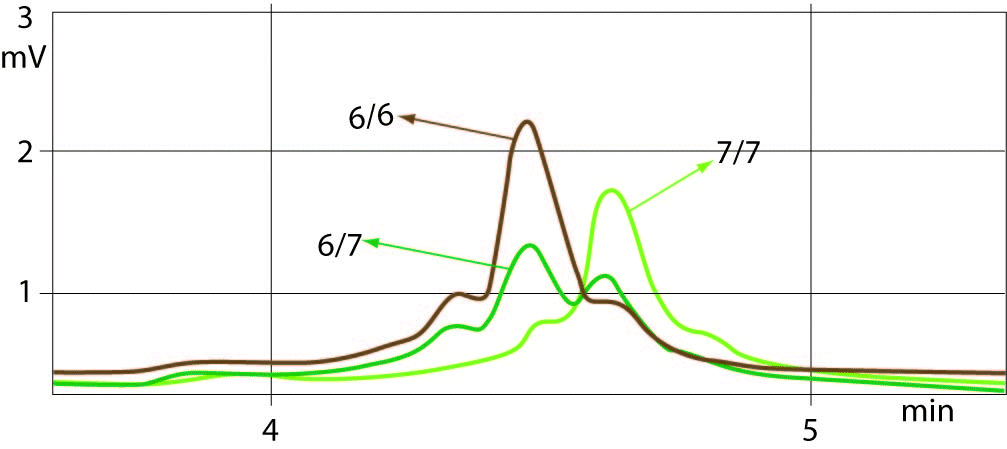
Figure 1. Example chromatograms from DHPLC sizing assay.
Allelic discrimination of the rare UGT1A1(TA)n polymorphism genotypes
For determination of the (TA)5/6 and (TA)77 or (TA)7/8 and (TA)7/7 genotypes of UGT1A1 promoter polymorphism the best quality controls for accurate genotyping were those with (TA)5/7 or (TA)6/8 genotypes, respectively. Allelic size discrimination of UGT1A1 promoter (TA)5/6, (TA)7/7and (TA)5/7 (a) and (TA)6/8, (TA)7/8 and (TA)7/7 (b) repeat genotypes using DHPLC assay is presented in Figure 2.
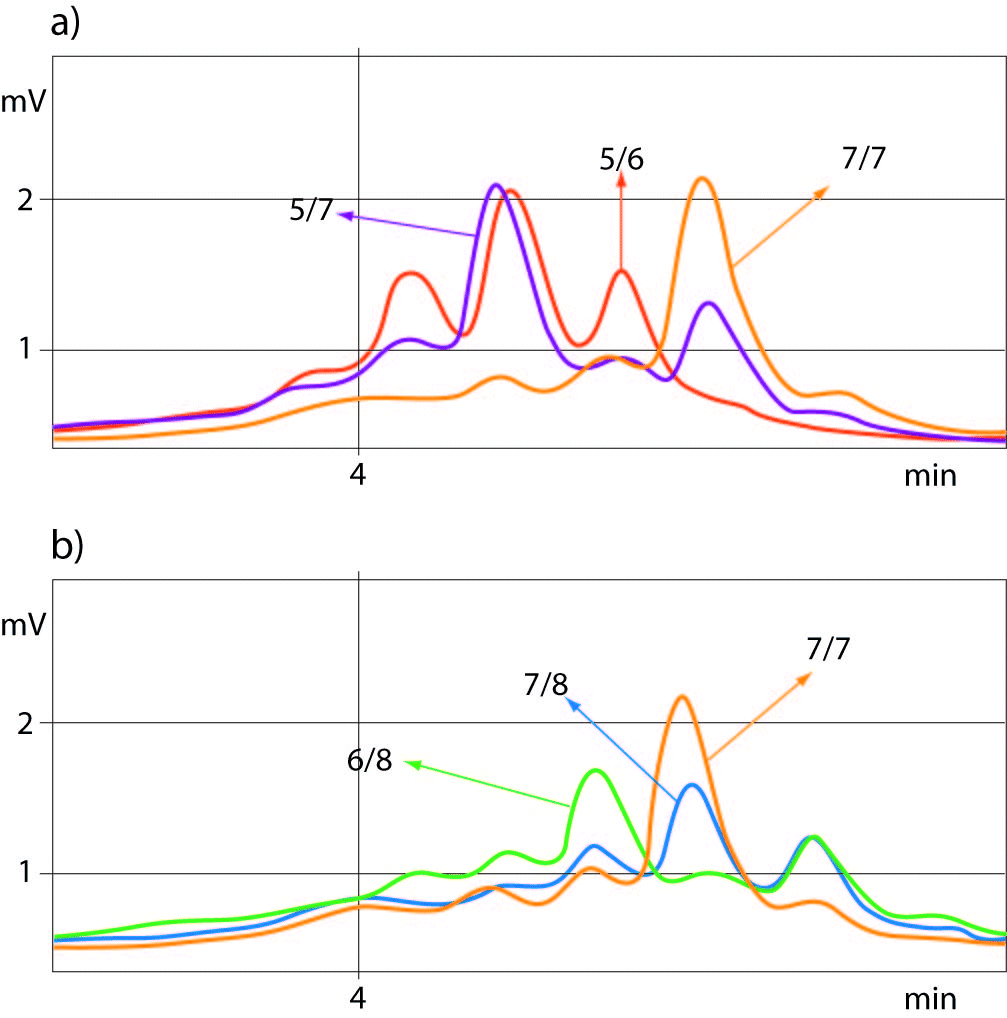
Figure 2. Allelic discrimination of UGT1A1-promoter (TA)5-7 (a) and (TA)6-8 (b) repeat genotypes using DHPLC sizing assay.
Samples with 5 TA and 8 TA repeat alleles eluted from the column showing the peaks at a retention time range of 4.076-4.104 min and 4.610 min, respectively.
The chromatograms showed double peaks in heterozygote samples with additional small peak eluted earlier, whereas the chromatograms of homozygous samples showed only one elution peak which was 2-times higher than in heterozygotes.
Discussion
The present study is the first to show the cost-effective, time-saving, accurate and rapid method for simultaneous genotyping of rare and common UGT1A1(TA)n promoter repeat genotypes in newly optimised conditions using sizing DHPLC method with the 100% genotype concordance rate with SSCP.
UGT1A1(TA)n genotyping of patients clinically suspected of having Gilbert syndrome can contribute to the confirmation of diagnosis, and may be important in individualisation of therapy for drugs that are metabolised by UGT1A1.
Routine methods, such as SSCP, high resolution polyacrylamide gel electrophoresis (PAGE), real time fluorescent PCR, pyrosequencing, and direct sequencing have been used for genotyping of UGT1A1(TA)n promoter repeat polymorphism (15) and also a DHPLC method has been recently introduced to many laboratories (17,19). Our experience is that the DHPLC technique is useful in polymorphism detection in heterogenous ethnic populations. Moreover, to date no studies have shown the assessment of both rare and common genotypes of the UGT1A1(TA)n polymorphism simultaneously using DHPLC method.
In this study, we compared the UGT1A1(TA)n repeat polymorphism genotyping in selected Slovenian patients using DHPLC and SSCP methods. Shorter PCR amplicons were prepared using primers specific for DHPLC closely flanking the UGT1A1 region with polymorphic (TA)5-8 repeats to achieve better separation of chromatogram peaks compared to longer PCR amplicons used for SSCP.
The exact lengths of UGT1A1(TA)n alleles were determined by running of UGT1A1(TA)n allelic sizing controls generated from heterozygous individuals. The lengths of UGT1A1(TA)n allelic sizing controls were characterized also by SSCP method. Using our DHPLC protocol, we were able to effectively genotype 57 selected samples. Also rare 7/8, 5/7, 5/6 and 6/8 TA repeats genotypes in 2, 1, 2 and 2 subjects were clearly genotyped, respectively, using the sizing method on DHPLC as well as SSCP.
The pattern of the elution profile for heterozygotes and homozygotes samples has shown that non-denaturing or sizing technique enables to distinguish between all genotypes of UGT1A1(TA)n polymorphism with no previous need for mixing of unknown homozygous PCR products with the known control homozygotes and denaturing/reannealing step before DHPLC run compared to denaturing technique (19). Due to avoidance of repeat analyses, the sizing method is more rapid and cost-effective and the labor requirements are thus reduced. Moreover, the study by Pirulli et al. (19) presented only the chromatogram peaks separation between 7/7, 6/6 and 6/7 TA repeats genotypes, whereas our study showed also the separation of other UGT1A1(TA)n genotypes occurring in our population. Moreover, Harraway et al. (17) separated PCR products based on molecular weight only. However, they showed peak separation between only 6-TA and 7-TA repeats using fully denaturing conditions on DHPLC. To date no study has shown the sizing separation of all repeats simultaneously ((TA)5/7, (TA)6/7, (TA)7/8) using only 50 °C on DHPLC. Due to using lower analysis temperature in our conditions rather than fully denaturing conditions (80%) (17) the run time of our sample analysis was then additionally shorter.
Our approach is simple, requiring only one PCR reaction with unlabeled primers and using lower-cost reagents than direct sequencing and real time fluorescence PCR.
All DHPLC-analyzed PCR samples were also run on the SSCP gels. Using DHPLC analysis, no sample was determined incorrectly (0%). Furthermore, the samples analysis DHPLC run takes only 6.6 min, therefore, operating times are shorter and labor cost lower than with SSCP method. Moreover, the SSCP assay for genotyping of either one or ten samples needs the same time-consuming pre-analysis preparation of gels and buffers, and detection occurs under the toxic conditions using silver staining of gels what is not needed for automated DHPLC analysis.
During our runs on DHPLC, the slight time shifting of individual chromatograms were seen, however the determination of true size of PCR fragment and then UGT1A1(TA)n genotype was still clear with no detection error. To overcome inappropriate sizing of individual alleles caused by slight time shifting of individual chromatograms during runs, the implementation of the quality controls with the known lengths of PCR amplicons were used in individual well of each running plate.
The critical points during sizing DHPLC analysis were the pressure in the column, temperature, flow rate, buffer purity, DNA quality as well as primer design since fragment lengths can also affect the DHPLC analysis accuracy.
The PCR amplification of the short polymorphic repeats generates PCR artifacts called shadow bands. Although their formation is not well understood, it has been proposed that the shadow bands are coming from stutter product as a result of AmpliTaq Gold polymerase slippage during DNA synthesis (22). In our experiment, the amount of shadow bands were higher in heterozygous samples, we assume then that they could represent the heteroduplexes formed by complementary DNA strands of two alleles hybridizing during the last PCR cycles. Despite present shadow bands the genotype detection was still clear.
In conclusion, we have developed a sizing DHPLC assay for simultaneous genotyping of rare and common UGT1A1(TA)n promoter polymorphism genotypes. We suggest this assay to be used as a rapid, automated, accurate, and cost-effective alternative method in routine clinical laboratories where the DHPLC machine is available as well as in large cohort genetics studies analyzing UGT1A1(TA)n promoter polymorphic repeats.
Acknowledgements
This work was supported by Grant P4-0127 provided by the Slovenian Research Agency.
The authors would like to thank Prof. Jana Lukač Bajalo, Assist. Danijela Furlan and Assist. Borut Bratanič for the samples.