Introduction
Genetics is the science of heredity and variation in living organisms, which investigates gene function, genome structure, chromatin organization, recombination rate, mutation processes, and evolutionary history, to provide a coherent understanding of the human genome and its complex relationship with human biology, physiology and disease. Genomics is instead typically defined as the intensive efforts to determine the entire DNA sequence of organisms and fine-scale genetic mapping efforts, including studies of intragenomic phenomena such as heterosis, epistasis, pleiotropy and other interactions between loci and alleles within the genome. Genetic testing was originally developed nearly twenty years ago and its original applications were limited to counseling and prenatal diagnosis of a few hereditary diseases. Due to the completion of the human genome project (HGP) (1), the subsequent International HapMap Project (which both have described genetic variations, mostly single nucleotide polymorphisms, among individuals and the patterns of variation across the genome) (2), and thanks to outstanding technological improvements, genetic testing is now widespread in clinical laboratories, supported by relatively inexpensive, fast, reliable high-throughput techniques. Indeed, genetic analysis has allowed humans to understand the molecular basis of several disorders, has produced a paradigm shift in diagnosis and clinical management of single gene disorders (e.g., hemophilias, thalassemias, etc.), has helped identify a variety of genes involved in most multifactorial prevailing pathologies (e.g., diabetes, cancer, venous thromboembolism, cardiovascular disease), has allowed the personalization of treatments and especially pharmacological therapies. However, it has also produced a paradox in the managed care of patients, in that most would perceive genetic testing as a panacea, with the assumption that each single genetic polymorphism is associated with a specific, individual phenotype and clinical picture. But, the truth is different, both for single gene disorders and, especially, for multifactorial pathologies. Some examples will be discussed below.
Monogenic disorders
Hemophilia A is a severe inherited bleeding disorder caused by X-linked recessive mutations resulting in deficiency of functional plasma factor VIII (FVIII). Its prevalence in the population (approximately 1 per 5000-10,000 males born) is due to a high mutation rate predominantly in male germ cells, namely, de novo somatic mutations in early embryogenesis (3). It is traditionally assumed that the most severe forms of FVIII deficiency (e.g., those with FVIII levels < 1%) are associated with a severely increased risk of bleeding from common injuries, which involves joints, muscles, gastrointestinal tract and brain. In the moderately severe forms of disease (e.g., FVIII levels between 1% and 5%), muscle and joint hemorrhages are also frequent, whereas digestive tract and cerebral hemorrhages might occur as well (4,5). The major problem in the managed care of hemophiliacs is personalization of prophylaxis and therapy. Although most scientists and clinicians rely on the traditional classification of disease (severe, moderate, mild), several lines of clinical evidence attest that the severity of symptoms varies widely among patients, also among those carrying the same (genetic) molecular defects. To further increase the complexity, no definite correlation exists between the clinical severity of hemophilia and the results of coagulation tests (activity of deficient factors, whole blood clotting time, thromboplastin screening test) either, since up to 10%-15% of patients with severe hemophilia (< 1% clotting factor activity) have relatively mild clinical disease, only rarely bleed and do not need prophylactic therapy (4,5). Moreover, it is also acknowledged that the onset of the development of antibodies to FVIII (called “inhibitors”) as the most severe complication in hemophiliacs might be heterogeneous (6). Although it has been reported that patients with severe hemophilia A and mutations predicting a null allele might develop inhibitors more frequently than those with missense mutations, the overall predictive value of genetic testing is questionable because 33% to 78% of hemophiliacs might be misclassified according to their individual genetic risk (7).
In face of these data, we would conclude that particular genotypes in hemophiliacs (as for other single gene disorders) are not always mirrored by identical biochemical and clinical phenotypes. First and foremost, the activity of FVIII in plasma (FVIII:C) is similar but not overlapping in patients with identical mutations because the half-life of FVIII in plasma is reportedly shorter in patients with blood group O and low von Willebrand factor (VWF) antigen levels (5). Accordingly, the current approach to correct the deficit by replacement therapy (recombinant FVIII or less frequently fresh frozen plasma) is driven by the clinics and the presenting plasma FVIII:C level rather than by the individual genotype. Apart from variable associations of the genotype with the levels of residual clotting factor activity, there is evidence to suggest that pharmacokinetics of administered clotting factor concentrates, the presence of prothrombotic markers (8), and variations in other coagulation proteins as assessed in tests of global hemostasis as well as the fibrinolytic system can affect both the presenting plasma FVIII:C and clinical severity of bleeding (4,5,9). Taken together, this evidence leads to the inevitable conclusion that the origin of the large heterogeneity of phenotypes in severe hemophilia is multifactorial and the contribution of genetic testing would therefore be only limited, so that the managed care of patients will be driven largely by clinical features and phenotypic laboratory tests such as factor activity and potentially thrombin generation assays in the near future. Even when predicting the clinical severity of hemophilia, the composite Hemophilia Severity Score (HSS) overlooks the genotype, and only includes the annual incidence of joint bleeds, World Federation of Hemophilia Orthopedic joint score, and annual factor consumption instead (10).
Polygenic disorders
Cardiovascular disease is the first cause of morbidity and mortality in Western countries and poses a substantial economic burden at the individual, institutional, and national levels. The accurate stratification of the risk and identification of the most appropriate triage for the patients are the leading goals for optimal clinical and economical management of this pathology (11). Besides the inherent phenotypic complexity, the presence of multiple causal factors is an additional challenge in investigating the genetic basis of cardiovascular disease (12). If fact, although a considerable overlap exists among various phenotypes, the underlying pathophysiology is usually different, since ischemic events (namely, acute myocardial infarction) often but not always results from rupture of a vulnerable atherosclerotic plaque, and the risk factors and their interactions to generate plaque vulnerability differ from those that influence coronary atherosclerotic burden (12). Accordingly, thousands of epidemiological research studies over past decades have led to the discovery of a strong but complex genetic background, which is mirrored by abnormal levels of several lipids and lipoproteins, abnormalities in glucose metabolism (especially diabetes), obesity, hypertension, inflammation, oxidative stress, and hypercoagulability (12).
In addition to its potential contribution to the diagnosis of monogenic disorders, genetic testing might also be of future value in multifactorial, polygenic pathologies such as the cardiovascular disease, in that it would allow the construction of personalized frameworks including risk stratification for primary or secondary prophylaxis, duration of anticoagulant therapy, and family studies. In such a situation, the rationale to perform genetic analyses is further highlighted by the prevalence of specific inherited factors (e.g., hypertension, hypercholesterolemia) and for the significant risk conferred by a combination of additional less frequent genetic defects. As previously mentioned, however, the combined effects of genotypes and/or haplotypes are so complicated and multifaceted that some patients might develop ischemic complications with no identifiable risks factors, whereas others will not despite carrying “high risk” genotypes. Taking for granted that the development of myocardial infarction is promoted by numerous risk factors, ranging from rather modifiable lifestyle habits (e.g., smoking, physical activity) to genetic predisposition, two decades of candidate gene analyses have failed as yet to explain the molecular basis for the genetic predisposition to this pathology (13). Moreover, it has yet to be proven whether all the allelic variants which have been linked to cardiovascular disease are really associated with functional effects that affect risk prediction, and whether the reported effect sizes of the associations achieve a sufficient degree of clinical significance (14).
Conceivably, the appropriate and discretionary use of genetic tests, under reasonable conditions and according to the clinical setting (screening or diagnosis), will currently provide great opportunities. Due to increasing cost constraints and their questionable utility for classifying risk, the estimated expense does not justify preventive screening in large populations of healthy people (15). However, individuals with a high familial risk might benefit from early detection strategies and biochemical and DNA-based testing, which can further refine their risk. Individuals with the highest familial risk might carry mendelian disorders associated with a large magnitude of risk for premature coronary artery disease, so that referral for genetic evaluation should be considered (16).
Pharmacogenetics
Coumarin derivatives are the mainstay of oral anticoagulant therapy (OAT) and comprise the most often used anticoagulant medication worldwide (warfarin). The managed care of patients on OAT is challenging, due to considerable variability in the dose-response, so that personalizing responses would represent a great opportunity in limiting the burden of adverse health outcomes. Several lines of evidence now attest that 50%-60% of the individual pharmacological response to warfarin and other anticoagulant drugs is genetically determined, influenced by polymorphisms in the genes encoding for vitamin K epoxide reductase (VKOR) and cytochrome P450 CYP2C9 (17). Pharmacogenetic testing has thereby been proposed as a valuable tool to aid prediction of dose response during initial anticoagulation and assess variability in dose maintenance. Once again, however, genetic testing does not provide a panacea, being influenced by several limitations that include the optimal composition of test panels, the limited information provided on the inter-individual variability, the lack of quality specifications, and, especially, the lack of definitive outcome analyses that unequivocally confirm the cost-effectiveness of this approach. The major hurdle comes from the predominant reliance of the individual pharmacological response on environmental variables and “random” events such as variation of vitamin K intake, gastrointestinal function, metabolic abnormalities that influence the synthesis of vitamin K-dependent coagulation, alcohol, and other drugs (18,19). It is therefore premature to conclude that routine genetic testing would carry an unequivocally favorable cost-to-benefit ratio, although the development and clinical validation of simple but comprehensive algorithms integrating the most informative gene polymorphisms with some demographic and clinical variables may provide a valuable tool in the individually managed care of patients on warfarin therapy.
Why and where the link is missing
Protein expression is regulated at several levels, including (a) transcriptional/posttranscriptional/pretranslational processes that impact the quantitative level of expression of mRNA, (b) translational processes that modulate the conversion of the mRNA blueprint into protein, and (c) posttranslational/degradative processes that impact the stability or half-life of the protein product. Several other potential processes exist but remain unknown, or their biological background has not been recognized. Other than gene transcription, additional aspects will impact on protein expression, including gene to gene interactions, comorbidities, biological variability, the environment, etc.
Lipoprotein(a) (Lp(a)) is a paradigmatic example of how all the variables can impact on the genotype to phenotype translation. The metabolism of this peculiar and highly atherogenic lipoprotein is reportedly under strict genetic regulation, in that more than 90% of its plasma concentration is influenced by quantitative and qualitative polymorphisms at the apolipoprotein(a) gene locus (20). One would expect, accordingly, that genetic testing can reliably predict the biochemical phenotype and the clinics that are ultimately plasma concentration and atherogenicity. Although this is partially true, there are some evident dissociations between genotype, phenotype and clinics instead (Figure 1). In fact, the presence of some interleukin 6 responsive elements in the sequence of the gene determines a substantial increase of the plasma concentration in response to inflammation (21). Likewise, sex hormones and physical activity also up-regulate gene transcription, renal failure can produce accumulation in plasma of Lp(a)-immunoreactive material because Lp(a) is also cleared by the kidneys, whereas some diets (e.g., alcohol consumption) and liver failure are associated with a substantial decrease of the plasma concentration since this organ is the main site of apolipoprotein(a) synthesis (21,22). Since the definitive Lp(a) particle is assembled in plasma by a covalent association between apolipoprotein(a) and the apolipoprotein B moiety of a low density lipoprotein, the plasma concentration of the latter strongly influences Lp(a) levels (21). Finally, concerning the atherogenicity of Lp(a), there is a debate as to whether high Lp(a) levels are compatible with longevity, so that this phenomenon would turn out as an apparent paradox (23). It has been consistently reported, however, that the atherogenicity of Lp(a) is tightly linked (subordinated) to the concomitant presence of additional risk factors (namely, hypercholesterolemia and hyperhomocysteinemia) that would ultimately unmask or enhance its atherogenic and anti-fibrinolytic potential (24).
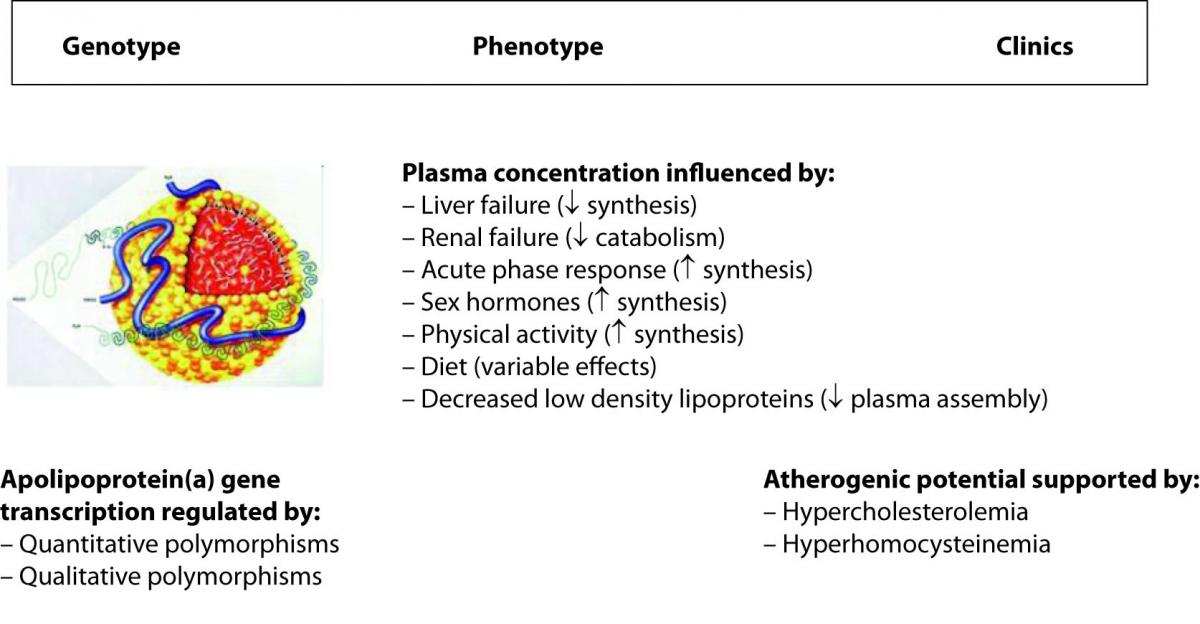
Figure 1. Major determinants of the plasma concentration of lipoprotein(a).
Concluding remarks
The major scope of translational medicine is seeking to close the gap between experimental research and clinical practice, and to develop and deliver new information that may assist prevention, diagnosis, and treatment of disease (25-27). With the completion of the HGP, many researchers are now looking at how genes and proteins interact to form other proteins. Unfortunately, the link between the genotype, the phenotype and the clinics is not always obvious, and it is even more challenging to address how much the link between gene and environment will impact on the managed care of patients. There is no doubt, however, that ongoing developments within predictive sciences which share the suffix -omics (genomics, proteomics, lipidomics, metabolomics, cytomics), will raise and stimulate a broad spectrum of research, allowing us to elucidate several steps at the genetic-subcellular-molecular-biochemical level that might enable us to overcome the traditional mechanistic approach to disease and to develop a personalized framework to stratify patients into different (even individual) subsets, each with common, but unique, characteristics (25-28). With the advent of high throughput microarray-based epigenetic technology (e.g., ChIP-on-chip and ChIP-seq), genetic testing will go beyond gene expression to explore gene regulation activity, thereby overcoming the inherent shortcomings of traditional genetic testing and providing the simultaneous measurement of the relative expression levels of thousands of individual genes.
Further hurdles come from financial, ethical, regulatory and practical barriers, and the last but not the least, the lack of effective communication between laboratory professionals and clinicians for the best possible use and interpretation of laboratory resources (29,30). Two ideal approaches can be advocated to investigate the multifaceted relationship between genetics, biochemistry and clinics. Genetic association (GA) studies offer a great opportunity for mapping causal genes with modest effects, but are limited because only a small number of genes can be studied at a time. In contrast, genome-wide association (GWA) studies will soon disclose a new scenario in the understanding of human biology, physiology and pathology (31). DNA microarray, or DNA chips are fabricated by high-speed robotics, allowing massively parallel gene expression and gene discovery studies. An experiment with a single DNA chip can provide researchers information on thousands of genes simultaneously, a dramatic increase in throughput, which is, however, very challenging to be handled from both sides (the laboratory and the clinic). Since GWA studies mostly rely on commercial single-nucleotide polymorphisms (SNPs) chips, for which a common evaluation criterion is global coverage of the genome (32), a major hurdle is again the cost-to-benefit ratio, in that it still remains prohibitively expensive and clinically questionable to genotype all the samples by a genome-wide genotyping array. To prevent the predictable and catastrophic scenario of a worldwide down-regulation of genetic testing (from pharmacies to “virtual”, uncontrolled facilities on the Web) (33), laboratory counseling will be foremost, instructing manufacturers to develop the most useful and predictive arrays, and guiding clinicians towards the most discretionary use of these powerful tools.
The concluding enigma is thereby: which is the real relationship between the genotype, phenotype and clinics? And, even more provocatively, would genotype analysis replace phenotypic testing in routine laboratory practice? There is no unequivocal answer to these questions as yet. Indeed, genetic testing has the great inherent potentials of identifying risk marriages, noninvasive prenatal fetal sex determination and preimplantation genetic diagnosis. It also helps predict, diagnose and treat pathologies, but it cannot be considered the “magic bullet” in the above mentioned pathologies as in other disorders because the predictivity of genetic testing gradually decreases throughout this translational process (Figure 2). Some important ethical issues should also be considered. At this point in time the link between genetic testing and clinics is labile for several pathologies, so that risk assessment might expose a large number of patients to false reassurances or excessive concerns. Hopefully, further scientific and technological advancements will help us understand the real role, usefulness and efficacy of genetic analyses, with the expectation that it will not yet replace phenotypic testing, which will remain a cornerstone of laboratory practice for several years to come.
Figure 2. The labile link between genotype, phenotype and clinics due to the decreasing predictivity of genetic testing.